First we will abstract the problem in the following way.
Suppose that we select k small intervals, each one
of length ![]() in the range 0 to 1.
Suppose that
in the range 0 to 1.
Suppose that ![]() is small enough so that we can
distribute the k intervals at random without
a significant danger of overlapping them.
We have now
is small enough so that we can
distribute the k intervals at random without
a significant danger of overlapping them.
We have now ![]() of the interval covered and
of the interval covered and
![]() uncovered.
The second step consists of choosing n random points
(
uncovered.
The second step consists of choosing n random points
(![]() )
in the unit interval.
We want to determine the probability that all the kintervals receive at least one of the n random
points.
)
in the unit interval.
We want to determine the probability that all the kintervals receive at least one of the n random
points.
Let
a1, a2, ..., ak, b be formal variables.
The probability we want to
compute is the one which includes
all the terms with each ai to the power 1 or higher.
These terms can be computed directly by computing which
are the coefficients independent of ai (ai to the
power 0) and subtracting this from the original
![]() .
To compute the probability, all the ai and b can
be set to 1. For example,
.
To compute the probability, all the ai and b can
be set to 1. For example,
 is
the probability that when selecting n points both intervals
of length
is
the probability that when selecting n points both intervals
of length
Although exact, this formula is extremely
ill conditioned
for computing these probabilities when ![]() is very
small and n and k are relatively large.
This cannot be ignored.
Assuming that
is very
small and n and k are relatively large.
This cannot be ignored.
Assuming that ![]() remains of moderate size
(neither too large nor too small), the following asymptotic
approximation can be used
remains of moderate size
(neither too large nor too small), the following asymptotic
approximation can be used
If we have k weights from a sampled protein and we match them against an unrelated digested protein which splits into n fragments, we can view the weights of these n fragments as random.
For example, suppose that we are given k=3weights of fragments: w1=441, w2=893 and
w3=1415 found by mass spectrometry from an
unknown protein.
Suppose that the theoretical digestion of a protein in the
database would give the weights v1=410,
v2=892, v3=925, v4=1218 and v5=1421.
If we require an exact match, none of the values
will match.
If we accept a tolerance radius r=1, then v2 is
within the range of w2,
![]() .
If we increase the tolerance to radius r=6, then two
weights will be within range,
.
If we increase the tolerance to radius r=6, then two
weights will be within range,
![]() and
and
![]() .
Finally we need to increase the tolerance to radius
r=31 to include all the given weights in ranges.
This information can be summarized by a table
.
Finally we need to increase the tolerance to radius
r=31 to include all the given weights in ranges.
This information can be summarized by a table
If the weights vi were from a totally unrelated protein, they could be viewed as random values in the given range. Lets assume, for the above example, that the weights were selected in the range 400 to 1500. We can now establish an equivalence between this example and the previously computed probabilities for each of the entries in the table. For example, the second line corresponds to hitting one interval with radius 1 out of 5 random choices. The third line corresponds to hitting two intervals each one within radius 6 out of 5 random points.
Before we compute these probabilities with the
above formulas, we need to consider the nature
of error in molecular weights.
These errors are not of an absolute magnitude,
rather they are relative to the mass we are
measuring.
I.e. an error of 1 for a mass of 1000 is equivalent
to an error of 10 for a mass of 10000.
Since our probability was derived on the assumption
that all the intervals were of the same length,
it is easiest to apply a transformation to the
masses, such that a constant relative error becomes
a constant absolute error.
In terms of our example, an error of 1 for the mass
of w1, 441 would be equivalent to an error
of
![]() for w3 with mass 1415.
A standard transformation, when we want to linearize
relative values, is to use the logarithms of the
measures.
So instead of working with the weights, we will
work with the logarithms of the weights.
A tolerance of
for w3 with mass 1415.
A standard transformation, when we want to linearize
relative values, is to use the logarithms of the
measures.
So instead of working with the weights, we will
work with the logarithms of the weights.
A tolerance of ![]() in the logarithms is
equivalent to a relative tolerance of
in the logarithms is
equivalent to a relative tolerance of
![]() in the relative values.
in the relative values.
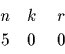
Finally, since our intervals in the theoretical derivation
were based on random variables distributed in (0,1),
we must make the relative errors relative to the size
of the entire interval, or divide them by
![]() where wmax and wmin are the
maximum and minimum weights that we will consider
in the sample.
where wmax and wmin are the
maximum and minimum weights that we will consider
in the sample.
In our example,
wmax=1500 and
wmin=400 so the
radius of the error for w1, v1 is
We have now all the ingredients for our algorithm.
For each protein in the database we compute its
digestion in, say, n fragments, and the weights
of these fragments.
For each searched weight wi we find the minimum
distance to one of the database weights vjand set
![]() .
Now we order the distances in increasing values
.
Now we order the distances in increasing values
 .
For distance di, i weights are within distance diof database weights.
For each di we compute
Pi,n,di.
The best match is considered the i for which
Pi,n,diis minimal.
This probability identifies the database sequence.
Finally we keep track of the m lowest probabilities
(m usually between 5 and 20).
.
For distance di, i weights are within distance diof database weights.
For each di we compute
Pi,n,di.
The best match is considered the i for which
Pi,n,diis minimal.
This probability identifies the database sequence.
Finally we keep track of the m lowest probabilities
(m usually between 5 and 20).
This function will be called SearchMassDb
and receives as parameters a set of weights, a function
which does the digestion, as shown earlier with
DigestTrypsin, and the value m.
> SearchMassDb := proc( weights:array(real), Digest:procedure,
> m:posint )
> description 'Search the database for similar mass profiles';
> if not type(DB,database) then
> error('a protein database must be loaded with ReadDb') fi;
create the output array (with maximum probability entries)> BestMatches := CreateArray( 1..m, [1] );sort the input weights and compute weight bounds
> w := sort(weights); > lw := length(w); > wmax := w[lw] * 1.02; > wmin := w[1] * 0.98; > d := CreateArray( 1..lw ); > IntLen := log(wmax) - log(wmin);for each sequence in the database do the digestion and compute the weights
> for e to DB[TotEntries] do > s := String(Sequence(Entry(e))); > v := Digest(s[1]); > v := sort(GetMolWeight(v));compute the di values
> j := 0; > lv := length(v); > for i to lw doposition
j such that

> for j from j to lv-1 while v[j+1] < w[i] do od; > if j > 0 then > d[i] := 2 * abs( log(w[i])-log(v[j]) ) / IntLen > else d[i] := 1 fi; > if j < lv then > di := 2 * abs( log(w[i])-log(v[j+1]) ) / IntLen; > if d[i] > di then d[i] := di fi > fi; > od;compute n, the number of weights between wmax and wmin
> n := 0; > for j to lv do > if v[j] >= wmin and v[j] <= wmax then n := n+1 fi od; > if n < 2 then next fi;sort the interval widths and compute the most rare event
> d := sort(d); > p := 1; > for k to lw do > if d[k] > 0 then > pk := ( 1 - exp(-n*d[k]) ) ^ k; > if pk < p then kmin := k; p := pk fi > fi > od;Instead of working with probabilities directly, we will work with the logarithm of these probabilities. To make these measures similar to the similarity scores of matches, we will compute
 .
This measure will now be comparable to
scores obtained
from alignments using the standard Dayhoff
matrices.
.
This measure will now be comparable to
scores obtained
from alignments using the standard Dayhoff
matrices. > sim := -10 * log10(p); > if sim > BestMatches[1,1] thennew smallest probability found, insert and reorder
BestMatches> BestMatches[1] := [sim,e,n,kmin]; > BestMatches := sort( BestMatches, x -> x[1] ); > fi > od; > BestMatches > end: