PROTEIN DIGESTION
Example protein:
YKVTLDQNRREGDIAKPNAED ...
will be broken into the following parts by Trypsin:
YK, VTLDQNR, R, EGDIAKPNAED ...
(splits after every R or K not followed by a P)
or if digested by Asp-N
YKVTLDQ, NRREGDIAKP, NAED...
(splits before every N)
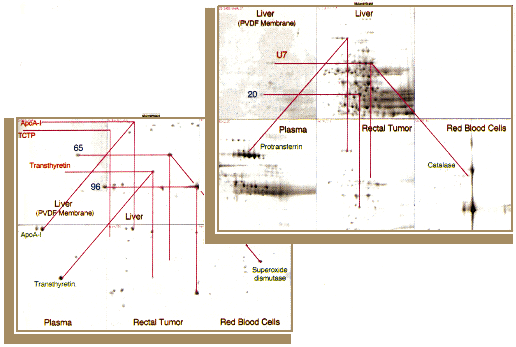
Figure 2. Computer match between silver stained 2-D PAGE patterns of liver (Fig. 1), plasma, red blood cell, rectal adenocarcinoma samples and an Amido Black-stained PVDF membrane pattern of liver sample (Fig. 6). This figure was obtained using all-spots, allareas, viewmod, modified automatch, showpairs, metal, gelsharper and showgroups programs of the Melanie/Elsie computer system [68, 71]. The color vectors link a few matched spots between the liver "master" picture, the PVDF membrane and the other type of samples. TCTP, translationally controlled tumor protein.
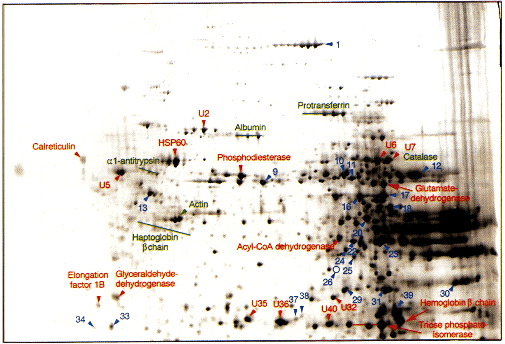
Figure 3 Enlargemement of the higher molecular weight area of Fig. 1. "U" indicates unknown sequence in Swiss-Prot database. The numbers provide a reference to Table 1. The green labels highlight proteins identified by gel comparison and the red labels those identified by N -terminal microsequencing. The blue labels highlight polypeptides which could not be N -terminally microsequenced either because of too low protein concentration or because of N -terminal blockage. HSP-60, heat shock protein 60.

Figure 4. Enlargement of the acidic and lower molecular weight area of Fig. 1. The yellow arrows highlight some spots which were unsuccessfully sequenced. We are currently attempting to get internal sequence information after in situ digestion, extraction and microbore reversed-phase HPLC. SRBP, serum retinol binding protein. Other details as in Fig. 3.
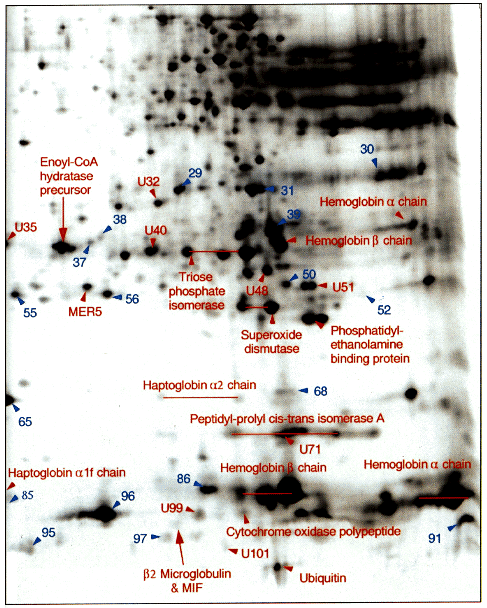
Figure 5. Enlargement of the basic area of Fig. 1. Labeling as in Fig. 3.
Question: Is it possible to predict composition from molecular weight?
Answer: NO.
Too many possible combinations
Mol. weight error
400
1000
2000
±0.5 386 9.780.723.528 1.577 x 1022 ±0.05 0 2.792.745.483 8.280 x 1020 ±0.005 0 391.021.208 4.608 x 1019 ±0.0005 0 173.920.080 1.979 x 1018
Number of sequences with given weight within error tolerance.
|
Alanine |
|
|
|
Arginine |
|
|
|
Asparagine |
|
|
|
Aspartic acid |
|
|
|
Cysteine |
|
|
|
Glutamine |
|
|
|
Glutamic acid |
|
|
|
Glycine |
|
|
|
Histidine |
|
|
|
Isoleucine |
|
|
|
Leucine |
|
|
|
Lysine |
|
|
|
Methionine |
|
|
|
Phenylalanine |
|
|
|
Proline |
|
|
|
Serine |
|
|
|
Threonine |
|
|
|
Tryptophan |
|
|
|
Tyrosine |
|
|
|
Valine |
|
|
- Reading of 2D gel electrophoretograms (2D gels)
Diagnosing diseases by 2D gel geometries
Identifying substances present/absent in healthy/sick cells.
- Determination of whether a protein is known or not before its sequencing
- In general: recognition of documented proteins from very small samples (fractions of pico-moles)
But our methods have to tolerate errors:
a) Recording error < 1%
b) Searched sequence not verbatim in databas (due to mutations)
c) Mutations may cause different digestions
d) Impurities in the sample and in the digester produce spurious data
e) Partial or incorrect digestion
f) Systematic error of apparatus
General Algorithm
Given a database D = {Di} where Di are vectors with ni values. Given a vector X with dimension k. Define dist (Di, X) = di a distance function. For a random vector Y of dimension n, compute
Prk,n,*E = Prob {dist(Y,x) <=p }.
*E (is going to be replaced by p due to that in HTML-files there does not exist a standard for greek-symbols).Select the database entries for which Prk,ni,di is lowest (rarest event).
Algorithm
- Given a set of molecular weights of the digested protein
- Digest theoretically each sequence in the database
- Compute the probability that a match of the given weights against the computed ones happens at random
- Record the lowest probabilities
Probability Foundations
Model: Balls thrown randomly in boxes of a given width.
1-unit total length
k-boxes each box of length p << 1
n-balls thrown at random
What is the probability that each of the k boxes has 1 or more balls?
Let a1, a2, .... aK,b be formal variables,
Gk, n, p = [a, p + a2p + .... akp + b(1-Kp)]n
is a generating expression of all the events of this experiment.
-Example:
the coefficient of a12bn-2 gives the probability that two balls fall in the first box and the rest outside all other boxes.
To compute probabilities, we set all formal variables to 1.
The answer to our problem is the sum of all terms a1e1 a2e2 .... akekbn-e where all the ei > 0.
-Example:
K = 2.
G2, n, p = [a1p + a2p + b(1-2p)]n
The coefficients in a1 to the power 0 sum up to [a2 p + b(1-2p)]n
(substitute a1 = 0), then
G*2,n,p = G2,n,p - (a2p + b(1-2p)]n
Are all the terms with a1e1 with e1 > 0.
Repeating for a2 and substituting the formal variables for 1, we find:
P2,n,p = 1 - 2(1-p)n + (1 - 2p)n
or in general:
Pk,n,p = sum[(bionomial (k,i)) (-1)i (1-ip )n] ~ (1-e-np )k[1 + O (knp 2e-np)]
-Example:
- from mass-spectrometer:
W1 441 W2 893 W3 1'415 - from theoretical digestion:
V1 410 V2 892 V3 925 V4 1'218 V5 1'421 -no exact match
-tolerance radius 1 - 1 match
-tolerance radius 6 - 2 matches
-tolerance radius 31 - 3 matches
Problem:
Errors should be considered relative, not absolute.
Solution:
work with logarithms of the weights, instead of the weights
|log wi - log vj| < p
1 - p ~ e-p ¾wi / vj ¾ ep ~ 1 + p
|(wi - vj) / vj | ¾ p
To normalize the interval to (0,1) we must divide the logarithms by (log wmax -log wmin) where wmax and wmin are the highest and lowest weights measured.
In our example:
wmax 1'500 wmin 400 normalized radius for w1, v1:
(log 441 - log 410) / (log 1'500 - log 400) = 0.055
for w2, v2:
(log 893 - log 892) / (log 1'500 - log 400) = 0.00084
and for w3,v3:
(log 1'415 - log 1'421) / (log 1'500 - log 400) = -0.0032
(The above are minimal radius, the corresponding intervals are twice this value.)
RESULTS FOR THE EXAMPLE:
|
n |
K |
p |
Prk, n, p |
Prk, n, p |
|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Further improvements
a) Multiple digestions (with different enzymes)
b) Systematic deviation (bias of the apparatus)
c) Using the global molecular weight to restrict the searching in each protein
d) DNA/RNA searching. It is possible to search a DNA/RNA database by converting each of the 3 frames of each sequence in each direction. This generates 6 times (or less) fragments than what is needed, but still works very well in practice.
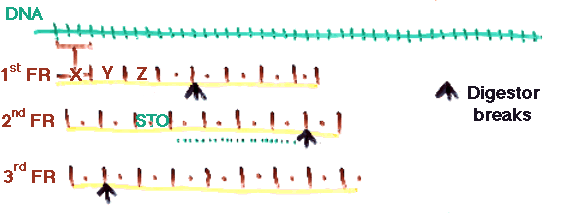
The expectation is that sufficient fragments will lie entirely in single exons.
e) Ambiguous amino acid modification. Some digestors produce ambiguous alterations of amino acids. This can be solved by digesting the database sequences in as many forms as it is possible.
f) Weight modification, e.g. deuteration. Any change which affects all amino acids is easy to resolve, by simply changing the weights during the database digestion.
Open Problems:
- Better than 0 (N) (linear) search of the database
- Better approximation to Prnkp
- Other measures of distance, (possibly overlapping boxes, etc.)
MassSearch: Searching SwissProt or EMBL by protein mass after digestion
ONE DIGESTER (TRYPSIN) 4 WEIGHTS
Score n k AC DE 0S 88.1 63 4 P18961; SERINE/THREONINE-PROTEIN KINASE YPK2/YKR2 (EC 2.7.1.). SACCHAROMYCES CEREVISIAE (BAKER'S YEAST). Unmatched weights: [776.0]. 67.4 41 3 P40943; ENDO-1,4-BETA-XYLANASE PRECURSOR (EC 3.2.1.8) (XYLANASE) (1,4-BETA-D-XYLAN XYLANOHYDROLASE). BACILLUS STEAROTHERMOPHILUS. Unmatched weights: [1353.0, 3739.0]. 64.4 6 2 P35898; POSSIBLE GUSTATORY RECEPTOR CLONE PTE45 (FRAGMENT). RATTUS NORVEGICUS (RAT). Unmatched weights: [776.0, 1353.0, 2232.O]. 61.7 35 3 P16823; HYPOTHETICAL PROTEIN UL71. HUMAN CYTOMEGALOVIRUS (STRAIN AD169). Unmatched weights: [2232.0, 3739.0]. 59.9 9 4 P35889; POSSIBLE GUSTATORY RECEPTOR CLONE PTE58 (FRAGMENT). RATTUS NORVEGICUS (RAT). Unmatched weights: [1353.0]. 59.8 10 2 P23268; OLFACTORY RECEPTOR-LIKE PROTEIN F12. RATTUS NORVEGICUS (RAT). Unmatched weights: [1353.0, 2232.0, 3739.0]. 59.0 57 3 P39194; !!!! ALU SUBFAMILY SQ WARNING ENTRY !!!! HOMO SAPIENS (HUMAN). Unmatched weights: [2232.0, 3739.0]. 58.4 46 3 P45861; HYPOTHETICAL ABC TRANSPORTER IN ACDA 5'REGION. BACILLUS SUBTILIS. Unmatched weights: [776.0, 3739.0]. 58.4 11 2 P03206; BZLF1 TRANS-ACTIVATOR PROTEIN (EB1) (ZEBRA). EPSTEIN-BARR VIRUS (STRAIN B95-8) (HUMAN HERPESVIRUS 4). Unmatched weights: [776.0, 2232.0, 3739.0]. 58.3 12 2 P28865; UL53 PROTEIN HOMOLOG (ORF1) (FRAGMENT). HERPES SIMPLEX VIRUS (TYPE 6 / STRAIN UGANDA-1102). Unmatched weights: [1353.0, 2232.0, 3739.0].TWO DIGESTERS (TRYPSIN, AspN) 6 WEIGHTS
Score n k n k AC DE 0S 137.4 63 3 21 3 P18961; SERINE/THREONINE-PROTEIN KINASE YPK2/YKR2 (EC 2.7.1.-). SACCHAROMYCES CEREVISIAE (BAKER'S YEAST). All weights matched. All weights matched. 74.1 37 2 16 1 P15278; FASCICLIN III PRECURSOR (FAS III). DROSOPHILA MELANOGASTER (FRUIT FLY). Unmatched weights: [3739.0]. Unmatched weights: [1785.0, 6509.0]. 70.0 22 2 8 2 P21348; FORMYLMETHANOFURAN--TETRAHYDROMETHANOPTERIN FORMYLTRANSFERASE (EC 2.3.1.101). METHANOBACTERIUM THERMOAUTOTROPHICUM. Unmatched weights: [3739.0]. Unmatched weights: [6509.0]. 68.4 16 2 9 1 P47614; HYPOTHETICAL PROTEIN MG374. MYCOPLASMA GENITALIUM. Unmatched weights: [2232.0]. Unmatched weights: [1785.0, 6509.0]. 64.4 6 2 4 1 P35898; POSSIBLE GUSTATORY RECEPTOR CLONE PTE45 (FRAGMENT). RATTUS NORVEGICUS (RAT). Unmatched weights: [2232. O]. Unmatched weights: [1785.0, 6509.0]. 64.4 30 1 9 2 P80468; ALCOHOL DEHYDROGENASE II (EC 1.1.1.1). STRUTHIO CAMELUS (OSTRICH). Unmatched weights: [309.3, 2232.0], Unmatched weights: [1389.0]. 64.3 53 2 13 2 P80313; T-COMPLEX PROTEIN 1, ETA SUBUNIT (TCP-1-ETA) (CCT-ETA). MUS MUSCULUS (MOUSE). Unmatched weights: [3739.0]. Unmatched weights: [6509.0]. 64.0 10 1 4 1 P36089; HYPOTHETICAL 16.7 KD PROTEIN IN NUP100-MSN4 INTERGENIC REGION. SACCHAROMYCES CEREVISIAE (BAKER'S YEAST). Unmatched weights: [2232.0, 3739.0]. Unmatched weights: [1389.0, 1785.0]. 63.1 39 2 17 1 P48570; HYPOTHETICAL 47.1 KD PROTEIN IN RPL41A-INH1 INTERGENIC REGION. SACCHAROMYCES CEREVISIAE (BAKER'S YEAST). Unmatched weights: [3739.0]. Unmatched weights: [1785.0, 6509.0]. 62.8 48 2 8 2 P07390; PET494 PROTEIN. SACCHAROMYCES CEREVISIAE (BAKER'S
ONE DIGESTER (TRYPSIN) TWO PROTEINS 3X2 WEIGHTS
Score n k n k AC DE 0S 110.1 13 3 23 2 P18961; SERINE/THREONINE-PROTEIN KINASE YPK2/YKR2 (EC 2.7.1.-). SACCHAROMYCES CEREVISIAE (BAKER'S YEAST). Unmatched weights: [1711.0]. Unmatched weights: [1306.3, 1456.0]. 104.1 15 2 20 2 P36026; UBIQUITIN CARBOXYL-TERMINAL HYDROLASE 11 (EC 3.1.2.15) (UBIQUITIN THIOLESTERASE 11) (UBIQUITIN-SPECIFIC PROCESSING PROTEASE 11) (DEUBIQUITINATING ENZYME 11). SACCHAROHYCES CEREVISIAE (BAKER'S YEAST). Unmatched weights: [2232.0, 3739.0]. Unmatched weights: [1785.0, 6509.0]. 81.7 7 3 12 1 P26484; FIXC PROTEIN. AZORHIZOBIUM CAULINODANS. Unmatched weights: [3739.0]. Unmatched weights: [1306.3, 1785.0, 6509.0]. 80.7 11 3 19 2 P06776; 3', 5'-CYCLIC-NUCLEOTIDE PHOSPHODIESTERASE 2 (EC 3.1.4. 17) (PDEASE 2) (HIGH AFFINITY CAMP PHOSPHODIESTERASE). SACCHAROMYCES CEREVISIAE (BAKER'S YEAST). Unmatched weights: [3739.0]. Unmatched weights: [1306.3, 1785.0]. 79.9 30 2 40 2 Q07518; RNA REPLICATION PROTEIN (CONTAINS: RNA-DIRECTED RNA POLYMERASE (EC 2.7.7.48) / PROBABLE HELICASE) (156 KD PROTEIN) (ORF 1). PLANTAGO ASIATICA MOSAIC POTEXVIRUS (P1AMV). Unmatched weights: [2020.0, 2232.0]. Unmatched weights: [1456.0, 6509.0]. 77.6 26 1 26 2 P29465; CHITIN SYNTHASE 3 (EC 2.4.1.16) (CHITIN-UDP ACETYL- GLUCOSAMINYL TRANSFERASE 3). SACCHAROMYCES CEREVISIAE (BAKER'S YEAST). Unmatched weights: [1711.0, 2020.0, 3739.0]. Unmatched weights: [1456.0, 6509.0]. 77.5 3 3 7 3 P14886; NIFY PROTEIN. AZOTOBACTER VINELANDII. Unmatched weights: [3739.0]. Unmatched weights: [1456.0].
SAMPLE RESULTS RECEIVED BY E-MAIL
MassSearch Trypsin: 1264.8, 1520.2, 955.9, 2487.0, 1094.1 AspN: 1624.4, 2961.4, 718.8, 716.9, 1890.0
The output of the above request is: Searching on SwissProt version 26. The sequences are printed in decreasing order of significance. Scores lower than 90 are probably not significant. For digester Trypsin, the fragment weights were: 1264.8 1520.2 955.9 2487.0 1094.1 For digester AspN, the fragment weights were: 1624.4 2961.4 718.8 716.9 1890.0 Score n k n k AC DE 0S 143.9 7 5 8 3 P02594; CALMODULIN. ELECTROPHORUS ELECTRICUS (ELECTRIC EEL). 143.9 7 5 8 3 P02593; CALMODULIN. HOMO SAPIENS (HUMAN), ORYCTOLAGUS CUNICULUS (RABBIT), BOS TAURUS (BOVINE), RATTUS NORVEGICUS (RAT), GALLUS GALLUS (CHICKEN), XENOPUS LAEVIS (AFRICAN CLAWED FROG), ONCORHYNCHUS SP. (SALMON) , AND ARBACIA PUNCTULATA (PUNCTUATE SEA URCHIN). 112.6 7 4 8 2 P21251; CALMODULIN. STICHOPUS JAPONICUS (SEA CUCUMBER). 94.7 21 3 22 2 P07265; MALTASE (EC 3.2.1.20). SACCHAROMYCES CARLSBERGENSIS (LAGER BEER YEAST). 94.2 7 4 8 2 P07181; CALMODULIN, DROSOPHILA MELANOGASTER (FRUIT FLY), LOCUSTA MIGRATORIA (MIGRATORY LOCUST), AND APLYSIA CALIFORNICA (CALIFORNIA SEA HARE).
e-mail RESULTS DNA SEARCHING RANDOM WEIGHTS
(NO SIGNIFICANT MATCH EXPECTED)
DNAMassSearch ApproxMass: 50000 Trypsin: M=83.092, 1264.8, 1520.2, 955.9, 2487.0, 1O94.1 AspN: Deuterated, 1624.4, 2961.4, 718.8, 716.9, 1890.0
The output of the above request is: Searching on EMBL version 35. The sequences are printed in decreasing order of significance. Scores lower than 100 are probably not significant. For digester Trypsin, the fragment weights were: 1264.8 1520.2 955.9 2487.0 1094.1 For digester AspN, the fragment weights were: 1624.4 2961.4 718.8 716.9 1890.0 Score n k n k AC DE 0S 100.3 85 2 46 3 M58040; Rat transferrin receptor mRNA, 3' end. Rattus norvegicus (rat) 100.1 99 2 49 3 Z18629; B. subtilis comF gene Bacillus subti1is 98.0 18 3 8 2 M37510; J04774; Human methylmalonyl CoA mutase (MUT) gene, exon 13. Homo sapiens (human) 93.4 42 4 30 3 M76493; H. contortus beta tubulin (tub8-9) mRNA, complete cds. Haemonchus contortus 93.4 21 3 9 3 M18356; Rat cytochrome P-450 (M-1) gene, exon 1. Rattus norvegicus (rat) 90.0 105 3 47 2 X65055; C.elegans cepgpC gene for P-glycoprotein C Caenorhabditis elegans (nematode)
DYNAMIC PROGRAMMING MASS SEARCH
SOLVES THE PROBLEM OF SEARCHING FOR A SUBSEQUENCE (FRAGMENT) GIVEN BY ITS PARTIAL WEIGHTS.
E.G. Frag:
K M E T E V A I E Y K S
1'427.6 (KM) E T E V A I E Y K S
1'168.2 (KME) T E V A I E Y K S
1'039.1 (KMET) E V A I E Y K S
0'938.0 etc. Then Find the Database sequence which matches those weights best. MASS Searching using Dynamic programming
M[1] := [1427.6, 1299.5, 1168.3, 1039.1]; (original)
M[1] := [1427.6, , 1168.3, 1039.1]; (test)
Matching against sequence entry 688: AC15_HUMAN: ACTIVATOR 1 140 KD SUBUNIT (REP LICATION FACTOR C LARGE SUBUNIT)
Simil: 27.79 MatchSimil: 18.35 MassSimil: 9.44
...kmEtevaieyks...
...KMETEVAIEYKS...
Matching against sequence entry 12657: FTSZ_STAAU: CELL DIVISION FTSZ PROTEIN.
Simil: 27.11 MatchSimil: 18.32 MassSimil: 8.79
...agmEkaikavvpaag...
...AGMEKAIKAVVPAAG...
Matching against sequence entry 47590: YMX2_YEAST: HYPOTHETICAL COX1/OXI3 INTRON 2 PROTEIN (AI2).:
Simil: 27.65 MatchSimil: 18.35 MassSimil: 9.31
...kmEehilrgvgr...
...KMEEHILRGVGR...
AVAILABILITY
- Algorithms implemented in Darwin. Darwin is distributed at no cost.
- Automatic e-mail server at ETH. All requests can be sent to
- WWW Web server. Information pages and same services as the e-mail server, but with a better user interface.
- Description/Math is in chapter 20 of "A Tutorial on Computational Biochemistry Using the Darwin System".
Zurich, 6th November 1997.

.gif)
.gif)
{kind=link}