For reasons both historical and algebraic, the mutation matrix is
transformed into a new matrix termed a Dayhoff matrix (in honour
of the first author, Margaret O. Dayhoff). The Dayhoff matrix, D,
is related to a 250-PAM mutation matrix by
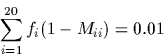
A 250-PAM distance corresponds to approximately ![]() identity
between two sequences (see §
identity
between two sequences (see §![[*]](cross_ref_motif.gif) for the proof). Many
believe this distance to be at the limit of our ability to detect
homology based on sequence data alone.
for the proof). Many
believe this distance to be at the limit of our ability to detect
homology based on sequence data alone.
Aligning sequences by dynamic programming using Dayhoff matrices is equivalent to finding the alignment which maximizes the probability that the two sequences evolved from a common ancestor as opposed to being random sequences. We are comparing two events:
The entries of the Dayhoff matrix are ten times the logarithm of the quotient of
these two probabilities.

Since dynamic programming maximizes the sum of the similarity measure, dynamic programming maximizes the sum of the logarithms or the product of these quotients. Therefore, dynamic programming finds the alignment which maximizes the probabiltiy of having evolved from a common ancestor (a maximum likelihood alignment) against the null hypothesis of being independent.