Recall, a 1-PAM mutation matrix measures a unit of evolution where the
expected change is exactly 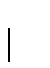 of the amino acids.
It is clear that a 2-PAM evolution will not produce a 2%
change since some changes
may mutate back to the original amino acid. (A 2-PAM evolution is
equivalent to two separate 1-PAM evolutionary units.)
The percentage identity, pi, is defined to be the number of identical
bases between two sequences in an alignment. It is is related to the
PAM distance, p, by
of the amino acids.
It is clear that a 2-PAM evolution will not produce a 2%
change since some changes
may mutate back to the original amino acid. (A 2-PAM evolution is
equivalent to two separate 1-PAM evolutionary units.)
The percentage identity, pi, is defined to be the number of identical
bases between two sequences in an alignment. It is is related to the
PAM distance, p, by
In the above formula we need both the mutation matrix and the
frequency vector. The frequency vector can be derived
from any mutation matrix.
Recall the symmetry relation:
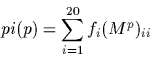
> PamToPerIdent := proc( pam:real, logPAM1:array(real,20,20) )
> description
> 'compute the percentage identity that a pam distance will leave';
> if pam < 0 then error('invalid percentage range') fi;
>
> num := den := 0;
> M := exp(pam*logPAM1);
> for i to 20 do
> num := num + M[i,1]*M[i,i]/M[1,i];
> den := den + M[i,1]/M[1,i]
> od;
> 100*num/den;
> end:
By definition, the following should be 99%> PamToPerIdent(1,logPAM1); 99.0000 > PamToPerIdent(250,logPAM1); 16.9051the classical PAM value gives about
 identity is needed to detect homology based
on sequence data alone.
identity is needed to detect homology based
on sequence data alone.
Computing the inverse function, PerIdentToPam, is somewhat more difficult. Since it is very unlikely that a closed form solution exists, we use Newton's method for solving equations.
Newton's method begins with an initial guess x0 and computes x1,
![]() using
using
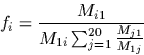
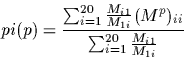
Consequently the derivative of the equation is
> PerIdentToPam := proc( pi:real, logPAM1:array(real,20,20) ) > description 'compute the PAM distance which results in the given > percentage identity';check the argument range and trivial case
> if pi <= 0 or pi > 100 then error('invalid percentage range') fi;
> if pi=100 then return(0) fi;
Compute the amino acid frequencies as described before> AF := CreateArray(1..20); > M := exp(logPAM1); > for i to 20 do AF[i] := M[i,1]/M[1,i] od; > AF := AF/sum(AF);Now check that the percentage identity is not less than its asymptotic value. That is, for PAM
 ,
the percentage identity
does not decrease to zero but to the percentage identity
of two random sequences.
This value is
,
the percentage identity
does not decrease to zero but to the percentage identity
of two random sequences.
This value is
> asy := AF*AF;
> if pi/100 <= asy then
> error('pi cannot be less than the asymptotic value',100*asy) fi;
>
For reasons which escape the scope of this tutorial,
the following is a good initial guess
for the PAM value.> pam := -100*ln(pi/100-asy);Next, the PAM value will be bound below by
lo
and above by hi. Setting the above bound to
5 times the initial guess is sufficient.> lo := 0; hi := 5*pam;We will now iterate until we reach the desired accuracy, i.e. the difference between the bounds is close to the machine epsilon.
> while hi-lo > hi*DBL_EPSILON*10 do > mp := exp(pam*logPAM1); > m1p := logPAM1*mp; > num := -pi/100; > den := 0; > for i to 20 do > num := num + AF[i]*mp[i,i]; > den := den + AF[i]*m1p[i,i] > od;Reset
lo or hi as appropriate> if num >= 0 then lo := pam else hi := pam fi; > incr := -num/den; > pam := pam + incr;If the increment squared is relatively less than the machine epsilon we can stop the iteration. Notice that when doing Newton's iteration, the relative error is proportional to the square of the previous relative error.
> if abs(incr)^2 < abs(pam)*DBL_EPSILON then break fi;If the value falls outside the bounds, then the Newton's value is not converging, so we just compute the midpoint of the bounds as the next guess.
> if pam <= lo or pam >= hi then pam := (lo+hi)/2 fi; > od; > pam > end:Again, by definition, the following should evaluate to 1.
> PerIdentToPam(99,logPAM1); 1.0000A
 identity corresponds to PAM 278.5236.
identity corresponds to PAM 278.5236.> PerIdentToPam(15,logPAM1); 278.5236