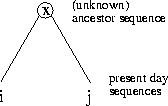
Definition 1.7
A 1-
PAM unit is the amount of evolution which will change,
on average,
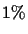
of the amino acids. In mathematical terms, this
is expressed as a matrix
M such that
where
fi is the frequency of the
ith amino acid.
Definition 1.8
The score of an
optimal pairwise alignment
OPA(
s1,
s2) of two
sequences
s1,
s2 is the score of an alignment with the
maximum score where a probabilistic scoring method
[
Dayhoff et al., 1978,
Gonnet et al., 1992] is used. We refer to a pairwise
alignment of two sequences
s1,
s2 with
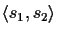
.