We will use DP over partially built multiple alignments and the C function will be derived from the probability of an EC. We call this aligning algorithm probabilistic dynamic programming.
More precisely, our DP algorithm works over two partial multiple alignments. A partial multiple alignment is a multiple alignment of all the sequences of a subtree of the EPT. Hence a partial multiple alignment is identified by an internal node and a direction in the EPT. E.g. node Y has as descendants the sequence 3 and the node W; node W has sequences 1 and 2 as descendants. The partial multiple alignment for node Y is:
1 FKQCCWNSLP____RGLSNVALVYQEFMAKCRGESENLQLVTALVINLPSMA 2 SMFRQCIWNSLS____HGLPETAPIYQPLKARCRGVSENLQLVTEIIINLPTLC 3 SLWCQCIKASLPLKVIRGTPEVAPLYDQLEQVCRSENQ____VSEIVAKFASLCFor each subtree X and its partial multiple alignment we also compute the TX and SX vectors. The C function for this algorithm will be derived from the probability of alignment of two slices of a multiple alignment under the same ancestor. In essence, this is exactly the same idea as in Dayhoff [6], extended so that we can compute the probabilities of alignments not just with two descendant sequences, but with two subtrees of descendants.
The C function is the logarithm of the quotient of probabilities, or
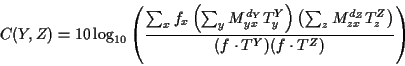
The cost of each computation of C is
![]() multiplications.
The cost of one probabilistic DP alignment is
multiplications.
The cost of one probabilistic DP alignment is
![]() and the cost of the entire process (including the computation
of S and T) is
and the cost of the entire process (including the computation
of S and T) is
![]() .
.
The alignment at the root of the EPT is the multiple alignment of all the sequences. Notice that for two sequences, this algorithm coincides exactly with DP using Dayhoff matrices, so this presents a continuous generalization of the 2-sequence algorithm.