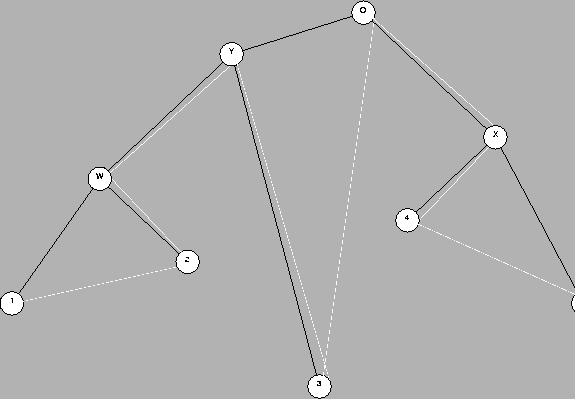
We will assume that we do not have any sequence information corresponding to the internal nodes of the phylogenetic tree. This is the normal situation. All the sequence information is at the leaves. Our phylogenetic trees are always binary (each internal node has degree 3), that is to say, every internal node corresponds to a splitting point in the phylogeny.
In our case, the leaves of the phylogenetic tree are the
sequences from present day species.
The internal nodes of the tree represent the points of
divergence where two different branches of evolution arose.
The root of the tree represents the nearest (latest) common ancestor
of all the species considered.
For example Figure 1
We are interested in trees with branches which measure amount of evolution in PAM units. We call this an evolutionary phylogenetic tree (EPT). There is no obvious constraint between the distances from the root to the leaves, as it is well known that similar proteins in different species may evolve at different rates.
In Figure 1, if lengths mean amount of evolution, we could say that 3 evolved more from O than 2 and 4. Since we assume that all these descended from a common ancestor, we can also say that 3 evolved more rapidly (mutated more rapidly) than 2 and 4, since in the same amount of time it evolved more.
When the ancestral sequences (O, Y, X and W) are not available for analysis, as it is usually the case, we have to infer their existence and location from the present-day sequences 1, 2, ..., 5. Phylogenetic trees derived only from the information on the leaves are normally called dendrograms.
The evolution from O to Ywill be modelled by a mutation matrix MdY, where dY is the PAM distance between O and Y; the evolution from O to 1 by MdO1 = MdY+dW+d1, etc. The exponent dij denotes the distance between node i and node j. di (with a single index) denotes the distance between node i and its parent.