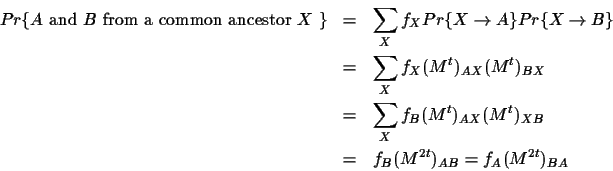Next: Monotonicity of scores and
Up: Optimal Scoring Matrices for
Previous: Model of Evolution
There are many tree construction methods based on PAM distances
[7,8,10,11,2]. PAM distances
are known to be consistent, that is, the more reliable the
distances are (usually when more data is available), the more
reliable the tree construction is. If an infinite amount of data
would be available, then the correct evolutionary tree could be
constructed [4,6]. A question usually
posed is: can scores derived from pairwise sequence alignments be
used for evolutionary tree construction? Are they consistent?
To compute scores for aligning sequences, the mutation matrix is
transformed into a matrix termed a Dayhoff matrix. The
Dayhoff matrix D is related to a 250-PAM mutation matrix by
The entries in the Dayhoff matrices are the logarithm of the
probability that the two amino acids evolved from a common
ancestor as opposed to being random sequences. This results from
the comparison of two events (see Figure 1):
- a)
- the two sequences are independent of each other. Hence an arbitrary position with amino acid A aligned to
another arbitrary position with amino acid B has the probability
equal to the product of the individual frequencies
- b)
- the two sequences have evolved from some common
ancestral sequence after some amount t of evolution.
Next: Monotonicity of scores and
Up: Optimal Scoring Matrices for
Previous: Model of Evolution
Chantal Korostensky
1999-07-14