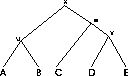
Definition 1.1
Given is a set of sequences
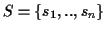
with
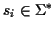
where
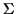
is a finite alphabet. A
Multiple
Sequence Alignment (in short, MSA) consists of a set of sequences
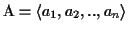
with
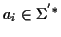
where
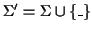
,
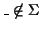
.
Furthermore
|
ai|=|
aj| for all
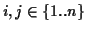
.
The sequence obtained by removing all gap
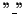
characters from
ai is equal to
si.
Definition 1.2
The character
or any contiguous sequence of
within an aligned sequence
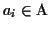
is called a
gap. A gap corresponds to an insertion or
deletion event (
indel). The length of a gap |
g| is the number of
.
Definition 1.4
Let
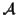
be the set of all possible MSAs that can be
generated for a given set of sequences
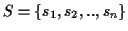
.
An
MSA scoring function
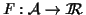
is a function, where a larger value
corresponds to a better alignment (otherwise the function can be multiplied by -1).
The
optimal MSA
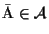
is an MSA such that
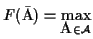
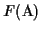
.