


Next: Discussion
Up: Simulation of Evolution
Previous: Simulation of Evolution
From a random evolutionary tree with 8 leaves a random MSA was
produced, that is, the original sequence was mutated and in this
case there were two indel events:
Generated random MSA:
---------------------------------------------
Score of the alignment (MPA): 1311.907
Maximum possible score (OPA): 1328.206
1 LETIDICKGCAALEYYRGPMIMRAMTSFRLDIKQQVGTTKACADATSNELTGAKLLHISDGQDTTIGQTVAIT
2 LELIDIEKGCAALEYNKGSMIMRAMTTFRLDLKDQVGSTAACADATKNKLTGAKLLHLSDGQESAMGQVVAIT
6 LHVIDERKRLEAARFNKGSVYLR___HVEIDLRTQVGSSPYAATVIKNVIKNTRPLKLCMGQELSLGMIVMLF
5 LHVIDERKRLPAARFNKGSVILK___HLEIDFQSSVGSNPRAATYVKNVIKGRKPLKLCDGQEISLGLIVCIW
7 LAVIEVRRGQVALEFNKGSVLLR___TLELDFQGQVGTPPRAAVYVKNVTKGAKPLHLVEGQEFNLGYVTCII
8 VHVADVTRGLTRLEFDKGSVVLR___HFELDFEGQAETNPRSSVYVKNVSQGVEPIHLTEWQEFNYGNVSCKI
3 LDVLDVTT___________IIIQ___TFRIDLQEQLGSNPASATYVKNILTGAKLLHLSEGEEYTMGHAVLIM
4 LDVLDVVT___________IIVV___TFRIDLQEQVGENPASASYVQDILTGAKLLHLSDGKEYTMGHVVAII
Figure 11:
Tree associated with example MSA
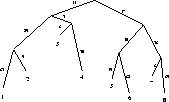 |
The score of the alignment is the MPA score introduced above. It
is the score derived from the pairwise alignments within the
MSA, in TSP order. In this example the order is 1, 2, 6, 5, 7, 8,
3, 4. You can easily verify that this tour is a circular tour of
the tree in Figure 11. The numbers on the edges of the
tree are PAM distances.
The maximum possible score, the
CSmax(S) score (see
Definition 2.4), is slightly better than the score
derived from the alignment (using MPA scores). This means that the
real alignment is not optimal, and the gaps could probably be
shifted a bit to either side to increase the score. The indel
events are one deletion event that happened in the ancestor of
sequence 3 and 4 and one insertion event that happened in the
ancestor of sequences 1 and 2.
Note that when the gaps are scored, each indel event is scored
only once. When sequence 8 is aligned against sequence 3, the gap
is scored. But when the alignment of sequences 3 and 4 is scored
in the MSA, the gap scores 0, because it has already been
accounted for, and you could actually simply remove the gap in the
MPA (see Definition 2.6), since they have exactly the
same length. Another observation is that the gaps form ``blocks'',
that is, the gaps that belong to the same evolutionary event are
not interrupted with sequences without gaps.
The sequences at the leaves are then fed to different algorithms.
None of the algorithms knows the correct tree or the correct MSA.
Sequences:
----------
1 LETIDICKGCAALEYYRGPMIMRAMTSFRLDIKQQVGTTKACADATSNELTGAKLLHISDGQDTTIGQTVAIT
2 LELIDIEKGCAALEYNKGSMIMRAMTTFRLDLKDQVGSTAACADATKNKLTGAKLLHLSDGQESAMGQVVAIT
3 LDVLDVTTIIIQTFRIDLQEQLGSNPASATYVKNILTGAKLLHLSEGEEYTMGHAVLIM
4 LDVLDVVTIIVVTFRIDLQEQVGENPASASYVQDILTGAKLLHLSDGKEYTMGHVVAII
5 LHVIDERKRLPAARFNKGSVILKHLEIDFQSSVGSNPRAATYVKNVIKGRKPLKLCDGQEISLGLIVCIW
6 LHVIDERKRLEAARFNKGSVYLRHVEIDLRTQVGSSPYAATVIKNVIKNTRPLKLCMGQELSLGMIVMLF
7 LAVIEVRRGQVALEFNKGSVLLRTLELDFQGQVGTPPRAAVYVKNVTKGAKPLHLVEGQEFNLGYVTCII
8 VHVADVTRGLTRLEFDKGSVVLRHFELDFEGQAETNPRSSVYVKNVSQGVEPIHLTEWQEFNYGNVSCKI
TSP ordering of sequences:
--------------------------
[1, 2, 6, 5, 7, 8, 3, 4, 1]
First, the sequences were fed to the MSA algorithm, which produces the
following output:
MSA:
---
Score of the alignment (MPA): 1305.050
Maximum possible score (OPA): 1328.206
1 LETIDICKGCAALEYYRGPMIMRAMTSFRLDIKQQVGTTKACADATSNELTGAKLLHISDGQDTTIGQTVAIT
2 LELIDIEKGCAALEYNKGSMIMRAMTTFRLDLKDQVGSTAACADATKNKLTGAKLLHLSDGQESAMGQVVAIT
6 LHVIDERKRLEAARFNKGSVYLR___HVEIDLRTQVGSSPYAATVIKNVIKNTRPLKLCMGQELSLGMIVMLF
5 LHVIDERKRLPAARFNKGSVILK___HLEIDFQSSVGSNPRAATYVKNVIKGRKPLKLCDGQEISLGLIVCIW
7 LAVIEVRRGQVALEFNKGSVLLR___TLELDFQGQVGTPPRAAVYVKNVTKGAKPLHLVEGQEFNLGYVTCII
8 VHVADVTRGLTRLEFDKGSVVLR___HFELDFEGQAETNPRSSVYVKNVSQGVEPIHLTEWQEFNYGNVSCKI
3 LDVLDV___________TTIIIQ___TFRIDLQEQLGSNPASATYVKNILTGAKLLHLSEGEEYTMGHAVLIM
4 LDVLDV___________VTIIVV___TFRIDLQEQVGENPASASYVQDILTGAKLLHLSDGKEYTMGHVVAII
The alignment looks very similar to the constructed MSA. The only
difference is that the gaps appear in different places, and the
score is slightly lower than the score of the constructed MSA. In
the simulation, the difference of the upper bound - the CS score
were noted (see tables). In this case the difference would be
23.156.
The next algorithm is the probabilistic model. In this case the
algorithm merges the two gaps:
Probabilistic model:
-------------------
Score of the alignment (MPA): 1305.080
Maximum possible score (OPA): 1328.206
1 LETIDICKGCAALEYYRGPMIMRAMTSFRLDIKQQVGTTKACADATSNELTGAKLLHISDGQDTTIGQTVAIT
2 LELIDIEKGCAALEYNKGSMIMRAMTTFRLDLKDQVGSTAACADATKNKLTGAKLLHLSDGQESAMGQVVAIT
6 LHVIDERKRLEAARFNKGS___VYLRHVEIDLRTQVGSSPYAATVIKNVIKNTRPLKLCMGQELSLGMIVMLF
5 LHVIDERKRLPAARFNKGS___VILKHLEIDFQSSVGSNPRAATYVKNVIKGRKPLKLCDGQEISLGLIVCIW
7 LAVIEVRRGQVALEFNKGS___VLLRTLELDFQGQVGTPPRAAVYVKNVTKGAKPLHLVEGQEFNLGYVTCII
8 VHVADVTRGLTRLEFDKGS___VVLRHFELDFEGQAETNPRSSVYVKNVSQGVEPIHLTEWQEFNYGNVSCKI
3 LDVLDVTT______________IIIQTFRIDLQEQLGSNPASATYVKNILTGAKLLHLSEGEEYTMGHAVLIM
4 LDVLDVVT______________IIVVTFRIDLQEQVGENPASASYVQDILTGAKLLHLSDGKEYTMGHVVAII
The last algorithm tested in this example is ClustalW. It looks like
the algorithm also found two indel events, but when you look closer
you can see that the gaps are shifted against each other. This means that the second
block of gaps are two indel events and not just one, which is the reason for the lower
score.
ClustalW:
---------
Score of the alignment (MPA): 1291.417
Maximum possible score (OPA): 1328.206
1 LETIDICKGCAALEYYRGPMIMRAMTSFRLDIKQQVGTTKACADATSNELTGAKLLHISDGQDTTIGQTVAIT
2 LELIDIEKGCAALEYNKGSMIMRAMTTFRLDLKDQVGSTAACADATKNKLTGAKLLHLSDGQESAMGQVVAIT
6 LHVIDERKRLEAARFNKGSVYLR___HVEIDLRTQVGSSPYAATVIKNVIKNTRPLKLCMGQELSLGMIVMLF
5 LHVIDERKRLPAARFNKGSVILK___HLEIDFQSSVGSNPRAATYVKNVIKGRKPLKLCDGQEISLGLIVCIW
7 LAVIEVRRGQVALEFNKGSVLLR___TLELDFQGQVGTPPRAAVYVKNVTKGAKPLHLVEGQEFNLGYVTCII
8 VHVADVTRGLTRLEFDKGSVVLR___HFELDFEGQAETNPRSSVYVKNVSQGVEPIHLTEWQEFNYGNVSCKI
3 LDVLDVTT___________III___QTFRIDLQEQLGSNPASATYVKNILTGAKLLHLSEGEEYTMGHAVLIM
4 LDVLDVVT___________IIV___VTFRIDLQEQVGENPASASYVQDILTGAKLLHLSDGKEYTMGHVVAII
Following is the result of a large simulation. For each tree type
hundreds of alignments were produced. This is the reason why the
scores have variances, because the upper bound is not the same for
each set of sequences, but lies in the same order, because similar
PAM distances were used and the same underlying tree structure.
Higher scores or smaller differences mean better alignments. The
rows are sorted into ascending order.
Figure 12 shows the result for balanced binary trees with
16 leaves. The length of the sequences is 300 amino acids and the
average edge distance is 30 PAM (so the maximum PAM distance
between two sequences is about 240 PAM).
Figure 12:
Comparison of different MSA methods: the CS score (second
column) is calculated using a TSP ordering. The upper bound is the CS score based
on the optimal pairwise alignment.
|
|
For this tree, MSA scored the best followed by PAS, ClustalW and
MAP. The alignments of MSA and PAS are slightly better than the
``real'' alignment. The reason for this is that the simulated MSAs
are not necessarily optimal. As a comparison the score of a bad
alignment (all the sequences aligned without deletions) was
calculated in the last row (Dummy). The tree on the right is an
example of a generated phylogenetic tree.
The next trees (Figure 13) are unbalanced with 30
sequences of length 300 and the average edge distance is 30 PAM
(so the maximum PAM distance is about 300). Only PAS, MAP and
ClustalW were able to compute the alignments (in a reasonable
time). In this case PAS did slightly better than ClustalW.
Figure 13:
Comparison of different MSA methods: the CS score (second
column) is calculated using a TSP ordering. The upper bound is the CS score based
on the optimal pairwise alignment.
|
|
The trees of the last table shown here (Figure 14) had 50
sequences with length 300 and an average edge length of 15 PAM. In
the simulation the PAS method was the best followed by ClustalW.
No other methods were able to calculate an MSA in a reasonable
time.
Figure 14:
Comparison of different MSA methods: the CS score (second
column) is calculated using a TSP ordering. The upper bound is the CS score based
on the optimal pairwise alignment.
|
|
These experiments show how the CS measure can be used. It also
shows that the upper bound
CSmax(S) is very close to the
actual score
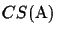 of the simulated MSA. Obviously the
ultimate evaluation of tools must be done with real rather than
simulated data.
of the simulated MSA. Obviously the
ultimate evaluation of tools must be done with real rather than
simulated data.
Next: Discussion
Up: Simulation of Evolution
Previous: Simulation of Evolution
Chantal Korostensky
1999-07-14


